Thermally Activated Delayed Fluorescence Material (TADF) Property Prediction¶
Note
- Before starting training and evaluation, please ensure the existence of property data files (.dat) and SMILES (smis.txt) data files, and modify
data_dirin the yaml configuration file to the property data file path andsim_dirto the SMILES data file path. - Before starting training and evaluation, please install
rdkitetc. For related dependencies, please executepip install -r requirements.txtto install.
| Pretrained Model | Metrics |
|---|---|
| Est.pdparams | loss(MAE): 0.045 |
| f.pdparams | loss(MAE): 0.036 |
| angle.pdparams | loss(MAE): 0.041 |
# Est Prediction:
cd TADF_Est
wget -c https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/Est/Est.dat https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/smis.txt
python Est.py mode=train
# f Prediction:
cd TADF_f
wget -c https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/f/f.dat https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/smis.txt
python f.py mode=train
# angle Prediction:
cd TADF_angle
wget -c https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/angle/angle.dat https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/smis.txt
python angle.py mode=train
# Est Evaluation:
cd TADF_Est
wget -c https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/Est/Est.dat https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/smis.txt
python Est.py mode=eval EVAL.pretrained_model_path=https://paddle-org.bj.bcebos.com/paddlescience/models/TADF/Est/Est_model.pdparams
# f Evaluation:
cd TADF_f
wget -c https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/f/f.dat https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/smis.txt
python f.py mode=eval EVAL.pretrained_model_path=https://paddle-org.bj.bcebos.com/paddlescience/models/TADF/f/f_model.pdparams
# angle Evaluation:
cd TADF_angle
wget -c https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/f/f.dat https://paddle-org.bj.bcebos.com/paddlescience/datasets/TADF/smis.txt
python angle.py mode=eval EVAL.pretrained_model_path=https://paddle-org.bj.bcebos.com/paddlescience/models/TADF/angle/angle_model.pdparams
1. Background Introduction¶
Organic Light-Emitting Diodes (OLEDs) have attracted widespread attention in advanced display and lighting technologies due to their advantages of high efficiency, flexible structure and low cost. In OLED devices, electrically injected carriers form singlet and triplet excitons in a ratio of 1:3. The theoretical limit of OLED luminous efficiency IQE constructed with pure fluorescent materials as luminescent materials is 25%. On the other hand, organometallic complex luminescent materials introduce rare metals (Ir, Pt, etc.) to bring strong spin-orbit coupling (SOC), which can convert singlet excitons into triplet excitons through intersystem crossing process, thereby utilizing triplet excitons to emit phosphorescence, and its IQE can reach 100%, but the high price of rare metals hinders its widespread use. Thermally Activated Delayed Fluorescence materials (TADF) provide new ideas for solving these problems and have attracted widespread attention. In TADF, triplets are converted into singlets and emit fluorescence through Reverse Intersystem Crossing process (RISC), thereby achieving 100% IQE, and the RISC process largely depends on the energy gap (\(\Delta Est\)) between the lowest singlet state (S1) and the lowest triplet state (T1). According to quantum mechanics theory, ΔEST is equivalent to twice the exchange integral between HOMO and LUMO. Therefore, a common design strategy for TADF molecules is to combine electron donor (D) and electron acceptor (A) with a significantly twisted dihedral angle to achieve significant separation of HOMO and LUMO in space. However, contrary to \(\Delta Est\), oscillator strength (\(f\)) requires a larger overlap integral between HOMO and LUMO, and the contradiction between the two needs to be further balanced.
2. Model Principle¶
The dataset is constructed through high-throughput computing, and molecular feature representation is realized through three links: molecular structure input, fingerprint feature extraction, and feature dimensionality reduction. Subsequently, the complex mapping relationship between molecular structure features and key TADF parameters is learned through multi-layer non-linear transformation, and finally end-to-end property prediction is realized.
3. Implementation of TADF Property Prediction Model¶
This example includes predictions of three properties: energy gap (\(\Delta Est\)), oscillator strength (\(f\)), and dihedral angle (\(angle\)) between electron donor and electron acceptor of chemical molecules. Next, taking dihedral angle \(angle\) as an example, we will explain how to implement the construction, training, testing and evaluation of the TADF property prediction model based on PaddleScience code. The directory structure of the case is as follows:
tadf/
├──TADF_angle/
│ ├── config/
│ │ └── angle.yaml
│ ├── angle_model.py
│ ├── angle.dat
│ ├── angle.py
│ └── smis.txt
├── TADF_Est/
│ └── ...
├── TADF_f/
│ └── ...
└── requirements.txt
3.1 Dataset Preparation¶
We choose 49 common acceptors and 50 donors to combine by single bond connection. By exhausting all possible combination sites, we obtained 44470 molecules. The initial structures of molecules were obtained by MMFF94 force field optimization. 5136 molecules were randomly extracted from 44470 molecules, ground state structure optimization was performed on 5136 molecules at B3LYP/6-31G(d) level, and excited state properties were calculated under ground state configuration using TDDFT method.
The data used in this case includes property data files (such as angle.dat) and SMILES data files (smis.txt). Each line of the molecular property data file is a molecular property; each line of smis.txt is a SMILES description of a molecule, that is, encoding the molecular structure into a linear string with a string of characters. Taking the first data as an example
Among them, lowercasec represents aromatic carbon, n represents aromatic nitrogen, and [nH] represents aromatic nitrogen with a hydrogen atom. Numbers 1, 2, 3 represent ring opening and closing marks: the first 1 opens a ring, and closes it when encountering the next 1.
After loading the property data file and SMILES data file line by line according to the information in the configuration file, first convert the SMILES description of the molecule into Morgan fingerprint through rdkit.Chem.rdFingerprintGenerator. Morgan fingerprint is a vectorized description of molecular structure, encoded as hash value through local topology, mapped to 2048-bit fingerprint bits. Subsequently, PCA is used to reduce the 2048 dimensions to dimensions where principal components retain 99% variance. Expressed in PaddleScience code as follows
3.2 Constraint Construction¶
This study uses supervised learning. According to the PaddleScience API structure description, the built-in SupervisedConstraint is used to construct supervised constraints. Expressed in PaddleScience code as follows
| examples/tadf/TADF_angle/angle_model.py | |
|---|---|
The second parameter of SupervisedConstraint indicates using mean squared error MSELoss as the loss function, and the third parameter indicates the name of the constraint condition, which is convenient for subsequent indexing.
3.3 Model Construction¶
For three prediction objects, the same deep neural network was designed. The network structure is a neural network containing two hidden layers. The first hidden layer contains 587 neurons, and the second hidden layer contains 256 neurons. Dropout is added between hidden layers. Taking \(angle\) prediction as an example, expressed in PaddleScience code as follows
| examples/tadf/TADF_angle/angle_model.py | |
|---|---|
3.4 Optimizer Construction¶
In the angle property prediction of this case, the trainer uses the Adam optimizer, the learning rate is set to 0.01, and weight_decay is set to 1e-5. Expressed in PaddleScience code as follows
| examples/tadf/TADF_angle/angle_model.py | |
|---|---|
3.5 Model Training¶
After completing the above settings, you only need to pass the instantiated objects to ppsci.solver.Solver in order, and then start training. Expressed in PaddleScience code as follows
| examples/tadf/TADF_angle/angle_model.py | |
|---|---|
4. Complete Code¶
| examples/tadf/TADF_angle/angle_model.py | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 | |
5. Result Display¶
The figure below shows the model prediction results for three properties: energy gap (\(\Delta Est\)), oscillator strength (\(f\)), and dihedral angle (\(angle\)) between electron donor and electron acceptor.
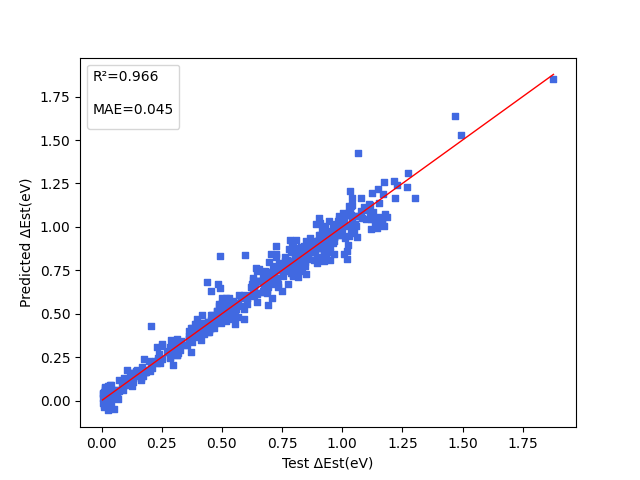
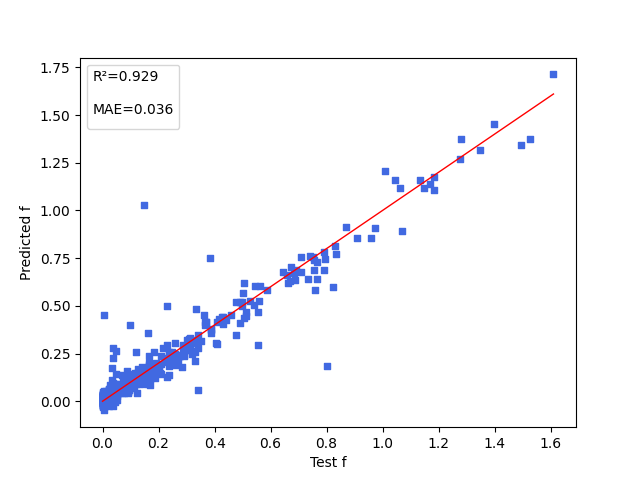
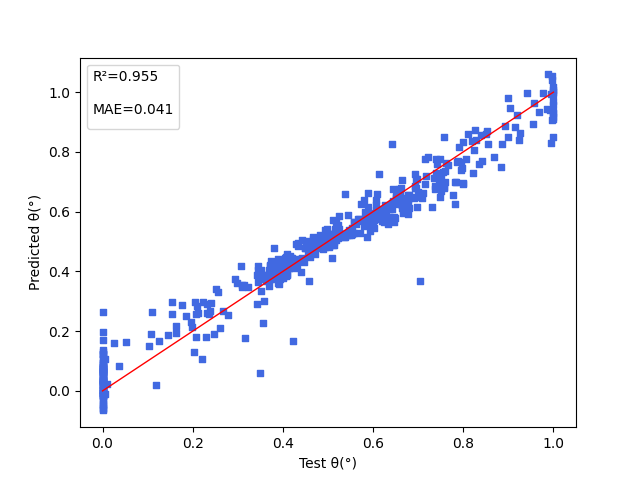
6. References¶
[1] Yufei Bu, Qian Peng*, Designing Promising Thermally Activated Delayed Fluroscence Emitters via Machine Learning-Assisted High-Throughput Virtual Screening. J. Phys. Chem. C. 2023. DOI: 10.1021/acs.jpcc.3c05337.